Optimizing the use of the extrapolation concept in pediatric investigational programs: Here are 4 key considerations.
Drug development in pediatrics continues to be a substrate for the application of innovative tools and techniques. When a new drug is approved in the United States, the European Union, or elsewhere for that matter, there is an expectation of a standard achieved for the evidence of effectiveness of the new molecule in question, as well as acceptable safety. This burden of evidence of effectiveness could depend on a particular disease or therapeutic area and could range from acute interventions to a more chronic nature. Thus, concepts of acute effect, maintenance of the effect and long-term outcomes come to mind. Three main issues emerge that relate to the disease itself, target engagement and pharmacology, and response to treatment. Collectively, they form the basis on which the principle of extrapolation can be assessed.
The concept of extrapolation is deeply embedded in region-specific legislation. These include the Best Pharmaceuticals for Children Act of 2002, and Pediatric Research Equity Act (PREA) of 2003 in the US, and the Paediatric Regulation of 2006 in the EU. Revisions to ICH E11 further deliberated the pediatric extrapolation concept. So, what is this extrapolation concept? In its most basic sense, extrapolation allows sponsors to decide on the level of clinical development that is needed to assess similarity between pediatric and adult populations, and streamline the manner in which pediatric studies are performed.
To enable the assessments underlying extrapolation, one needs to better understand the disease in question, the pharmacology of the drug, as well as the clinical response to treatment. An assessment on the potential sources of differences is also vital, as there might be underlying drug metabolizing enzymes that could affect the way the drug is metabolized as age decreases. Finally, methods to understand similarity between adults and pediatric subjects are often subject to extensive debate.
The case in point story of belimumab (Benlysta) deserves a mention in any blog considering the extrapolation concept. Belimumab was approved by the FDA in adults in 2011. Part of the conditions of approval included a PREA commitment to undertake a pediatric phase 2 study in SLE. Whereas the initial trial needed a sample size of 100 pediatric subjects to be enrolled, due to recruitment challenges, the trial did not meet this sample size. FDA reviewers applied a Bayesian prior post-hoc to justify a lower sample size and released the sponsor from PMR. While considerable debate lingers on in the scientific literature on the FDA’s approach, it does create an opportunity for novel trial designs prospectively within pediatric studies to reduce the burden of patient trial access.
In this blog, we reflect on a few key considerations that are central in the optimal use of the pediatric extrapolation concept.
#1: Understanding pharmacology and ontogeny of drug metabolizing enzymesOne of the first questions that is typically asked is whether the pharmacokinetics in pediatric subjects might differ when compared with adults. This mainly applies to small molecules, whose metabolism and elimination may be dependent on drug metabolizing enzymes/or transporters. The answer lies in developmental ontogeny of drug metabolizing enzymes and/or transporters. It is possible that during the lower developmental age, some of these pathways may not have matured and therefore, the compound may be metabolized to a different extent than one might think in adults. Alterations in clearance could predispose pediatric subjects to differences in pharmacokinetics, importantly when clearance is impaired and thus exposures are likely to be higher in pediatrics when compared with adults. It is imperative that scrutiny be given to the principal pathways of drug metabolism and transporters for the compound in question, and whether there are data available on the ontogeny.
Both phase 1 and phase 2 enzymes are subject to age-dependent changes in expression and functional capability for biotransformation. For example, CYP2D6 generally reaches adult competence by 3-5 years of age (1). Thus, compounds whose metabolism is principally by CYP2D6 must take this into consideration if they are being studied in pediatric age groups <5 years. One way to mitigate against these potential differences in pharmacokinetics is to consider a PK-lead in cohort to understand how different PK in pediatrics might be before proceeding into a larger pediatric study. Similarly, adult competence of CYP1A2 activity was attained by 3 months of postnatal age (2). UGT1A9 activity does not appear to be present at birth but reaches adult levels by 4 months of age (3). Of the transporters described in the literature, OCT1 expression shows the most significant age-dependent change, where approximately 5-fold increase in protein expression was observed between neonates vs. adults (4). For a good summary of ontogeny in transporters refer to the review by Brouwer 2015 (4).
Much like the metabolism data, emphasis should also be given to the developmental expression of the biomarkers of target engagement for the compound, since changes in response to treatment may be evident between pediatrics and adults. This is further elaborated in the section below on clinical response to treatment (see #4).
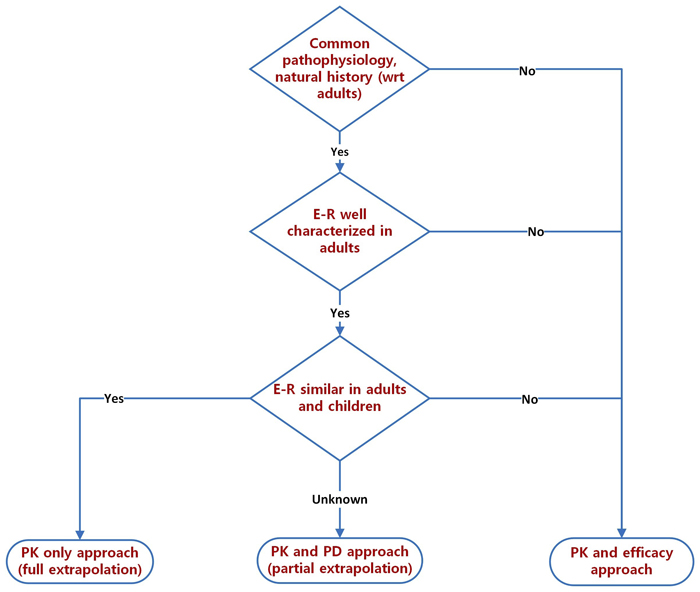
#2: Exposure/response considerationsOne can extrapolate the efficacy from studies conducted in adults to the pediatric population using the extrapolation framework. Two options are usually considered in this regard, a full extrapolation or a partial extrapolation. Central to this framework is a conceptual consideration of similarity in exposure and response between pediatric and adult subjects. Oftentimes, the most common path to dose selection in pediatrics is based on exposures matched from adults at clinically relevant doses. The so-called exposure-matching strategy is one that elicits the most interesting debates within pediatric investigational programs. This approach allows one to identify an unequivocal exposure-response relationship in an adult population to predict the dose and dosing regimen in the pediatric population, assuming that there are no inherent uncertainties that complicate such a relationship (e.g., developmental ontogeny). Needless to say, exposure/response relationships form the cornerstone of the extrapolation strategy in any successful pediatric development program.
A successful exposure/response strategy is one where there is a bonafide approach to dose selection in pediatrics based on the weight of evidence from adult data, design of PK sampling strategies to mitigate against potential differences in PK in pediatrics vs adults including clinical trial design considerations for lead-in PK cohorts, as well as methodology-based strategy that provides the basis of assessing comparability or similarity between adults and pediatrics at completion of pediatric trials (Table 1). Regarding the latter point, regulators typically expect acceptance margins to be prospectively defined.
Table 1: Key ingredients of a well-defined exposures/response strategy
| Step 1 Define target exposure range | Step 2 Select dose in pediatric study | Step 3 Show comparability |
| Perform exposure/response relationships with adult studies | Use adult population PK model to predict PK in pediatrics | Assess exposure/response in pediatric study |
| Define efficacious exposures for adult population | Ensure doses selected are matched to adult-equivalent exposures | Confirm pediatric exposure/response is similar to adult exposure/response |
It is instructive to consider regulatory opinions in guidance documents in this regard. The EU reflection paper on extrapolation, for example, emphasizes that modeling approaches in lieu of PK or PK/PD studies will only be considered acceptable when there is high confidence in physiology, ontogeny and compound related attributes.
#3: Disease considerationsFor successful extrapolation, it is necessary to sequentially ensure that the disease population, study design and response measures are aligned between the adult and pediatric age groups. Key disease considerations for extrapolation of adult to pediatric population are: pathophysiology, natural history and disease severity/presentation.
Only when disease pathophysiology and natural history are similar, can an extrapolation (either full [PK comparison only] or partial [PK/PD comparison]) be considered. The presentation of the disease in children, however, may vary, with age being a significant covariate. The younger the child, the more the pediatric disease likely differs from adults. Many adult diseases may not have an exact pediatric equivalent, especially in the youngest children. For example, sarcoidosis hardly occurs in very young children, with the few childhood cases being reported mostly in adolescents (5, 6). In slightly older (pre-adolescent) children, diseases start to show more similarities to adult disease, albeit with variations in severity. A typical example is type 1 diabetes mellitus where earlier age of onset generally presages more severe disease (7). As one gets to adolescence, diseases generally tend to resemble adult disease with just minor variations.
These age dependent changes have a significant bearing on extrapolation. It is easiest to extrapolate to adolescents from adults. Indeed, should adolescent disease be indistinguishable from adults, they may be included as part of adult studies, thereby obviating the need for any extrapolation! As one gets to pre-adolescent children, a precondition for extrapolation is the need to ensure a reasonable similarity of disease presentation/severity with adults. This will give a reasonable ability to show similarity in the PK/PD relationship, despite being unable to control for factors such as differing standards of care (SoC). In the youngest age groups, differences in disease presentation may lead to a situation where one can only include subsets of the pediatric population that have similarities with adults.
Despite the incremental changes with decreasing age that restrict the population to which extrapolation could be utilized, typically, regulators require that appropriate representation be given to all applicable age ranges – to ensure all applicable children have the opportunity to benefit from the drug (8).
#4: Trial designs and clinical response to treatmentThe key principle of extrapolation is to make maximum use of available adult information to make inferences about efficacy of the drug in children. This avoids the need for a formal efficacy related hypothesis testing, which is an expensive, effort and time consuming, and, occasionally, unfeasible exercise. To ensure the maximum number of children benefit from this exercise, pediatric studies are designed to include all applicable pediatric populations. Given such physiologically disparate populations, there may be a propensity to divide the pediatric population into many age ranges. A flexible approach is preferable with respect to age classification, since dividing into many age groups may needlessly increase the number of patients required and adds to study complexity.
A key design consideration during pediatric extrapolation is to ensure that there is a robust reference, i.e., reasonably contemporary adult population (against which pediatric PK/PD will be compared for similarity). Factors influencing the disease population in children are summarized in the section above. Once the populations are ascertained to be reasonably similar, study design elements are compared. Ideally, the pediatric study design should mimic the adult Phase 3 design to allow robust extrapolation. Should this not be possible, the pediatric study should have extrapolatable period(s) from adult (preferably Phase 3) studies. For example, the adult Phase 3 study may have a randomized withdrawal design (typical in many autoimmune conditions). If the pediatric study is a treat-through design, the period prior to randomized withdrawal in adults is extrapolatable. This could be supplemented with available data from adult (treat-through) study data from earlier (e.g., Phase 2) studies.
A placebo control is increasingly not being required in pediatric studies. This is mainly due to ethical (disease severity may preclude placebo control in children) and feasibility considerations (parents may not want to enroll their children when they are not sure of receiving potentially efficacious active treatment). To address this issue, an indirect placebo comparison is made – where adult active drug data is used as a bridge to evaluate pediatric efficacy in relation to adult placebo. Supportive descriptive comparisons of efficacy (over and above the PK/PD extrapolation) combine adult placebo data with pediatric active arm data for an integrated efficacy evaluation.
Generally, it is useful to utilize more than one active study arm in pediatric studies. This allows randomization and double-blinding of study arms and a greater range of PK exposures. Randomization and blinding allow a more unbiased efficacy and safety evaluation. Greater range of PK exposures are useful for the exposure-response analyses. However, single arm (by definition open-label) could be considered if the PK is well characterized, target exposure in children can be well defined and response measures are objective in nature. If populations include very young children, safety is critical and a single arm may be the only feasible option, since immature physiologic processes (refer #1 above) for drug metabolism may not allow prediction of PK.
Once the disease population and study design factors are finalized, the concluding step is to granulate the response measures. Adult registrational studies typically use objective endpoints or well characterized, validated endpoints which may include subjective components. Depending on the disease condition, the endpoints may also incorporate validated biomarkers as surrogates for clinical efficacy. In adolescents, adult endpoints are generally applicable and may even have been validated. In pre-adolescent children, while it may still be possible to apply adult endpoints, they may be sub-optimal to use compared to pediatric-specific endpoints that are better reflective of response. Insofar as extrapolation is concerned, it hinges on adult response measures. Therefore, every effort should be made to collect adult-specific response measures in pediatric studies. Pediatric-specific measures may be used to supplement the adult measures.
In the very young children, adult endpoints may not be used or may not be applicable (e.g. adult response measures are less relevant compared to pediatric-specific response measures for assessing multiple sclerosis in young children) (9). Oftentimes, one may not even have response measures defined in such young age groups. In such situations, extrapolation from adults may not be possible. However, disease response could still be evaluated using available response measures by analyzing changes from the baseline to elucidate drug effects.
Even after addressing all the above factors, there may still be knowledge gaps. However, it is anticipated that these will be relatively minor and may not carry enough weight to alter the overall comparison of exposure-response between adults and children.
In summary, pediatric development program represents an intriguing opportunity for the integrative scientist in ensuring the principles of extrapolation are optimally considered. Key design improvements are possible with model-informed approaches as they relate to endpoint selection, pediatric dose selection decision, design of the optimal clinical pharmacology and clinical studies, as well as ensuring model-based similarity in exposure/response relationships between adult and pediatric subjects are established.
For an overview on pediatric drug development, please read our blog Pediatric Drug Development: Four Critical Considerations
by Rajesh Krishna and Abhijeeth Chandrasekaran
Rajesh Krishna, PhD, is Senior Director, Integrated Drug Development at Certara, NJ, USAAbhijeeth Chandrasekaran, MD, is Director, Clinical Development at RxMD
References- Leeder JS, Kearns GL. Pharmacogenetics in pediatrics: implications for practice. Pediatr Clin North Am 1997;44:55-77.
- Tateishi T et al. Developmental changes in urinary elimination of theophylline and its metabolites in pediatric patients. Pediatr Res 1999;45:66-70.
- Strassburg CP et al . Developmental aspects of human hepatic drug glucuronidation in young children and adults. Gut 2002; 50:259–265.
- Brouwer KL et al. Human ontogeny of drug transporters. Review and recommendations of the pediatric transporter working group. Clin Pharmacol Ther 2015;98(3):266-87. doi: 10.1002/cpt.176.
- Pattishall EN et al. Childhood sarcoidosis. J Pediatr. 1986;108(2):169-77. doi: 10.1016/s0022-3476(86)80978-7.
- Kendig EL: Sarcoidosis in children. Personal observations on age distribution. Pediatric Pulmonol. 1989, 6: 69-70. 10.1002/ppul.1950060202.
- Amador-Patarroyo MJ, Rodriguez-Rodriguez A, Montoya-Ortiz G. How does age at onset influence the outcome of autoimmune diseases? Autoimmune Dis. 2012;2012:251730. doi: 10.1155/2012/251730. Epub 2011 Dec 13.
- ICH E11(R1) guideline on clinical investigation of medicinal products in the pediatric population. 1 September 2017. EMA/CPMP/ICH/2711/1999. Committee for Human Medicinal Products.
- Santoro JD et al., US Network of Pediatric MS Centers. Pediatric Multiple Sclerosis Severity Score in a large US cohort. Neurology. 2020;95(13):e1844-e1853. doi: 10.1212/WNL.0000000000010414. Epub 2020 Jul 20.
Suggested reading
- Ethical considerations for clinical trials on medicinal products conducted with minors. Recommendations of the expert group on clinical trials for the implementation of Regulation (EU) No 536/2014 on clinical trials on medicinal products for human use. Revision 1, 18 September 2017
- ICH Topic E11. Clinical Investigation of Medicinal Products in the Paediatric Population. January 2001. CPMP/ICH/2711/99.
- FDA Guidance for Industry. December 2014. General Clinical Pharmacology Considerations for Pediatric Studies for Drugs and Biological Products.
For more blogs, please visit: https://www.rxmd.com/insights